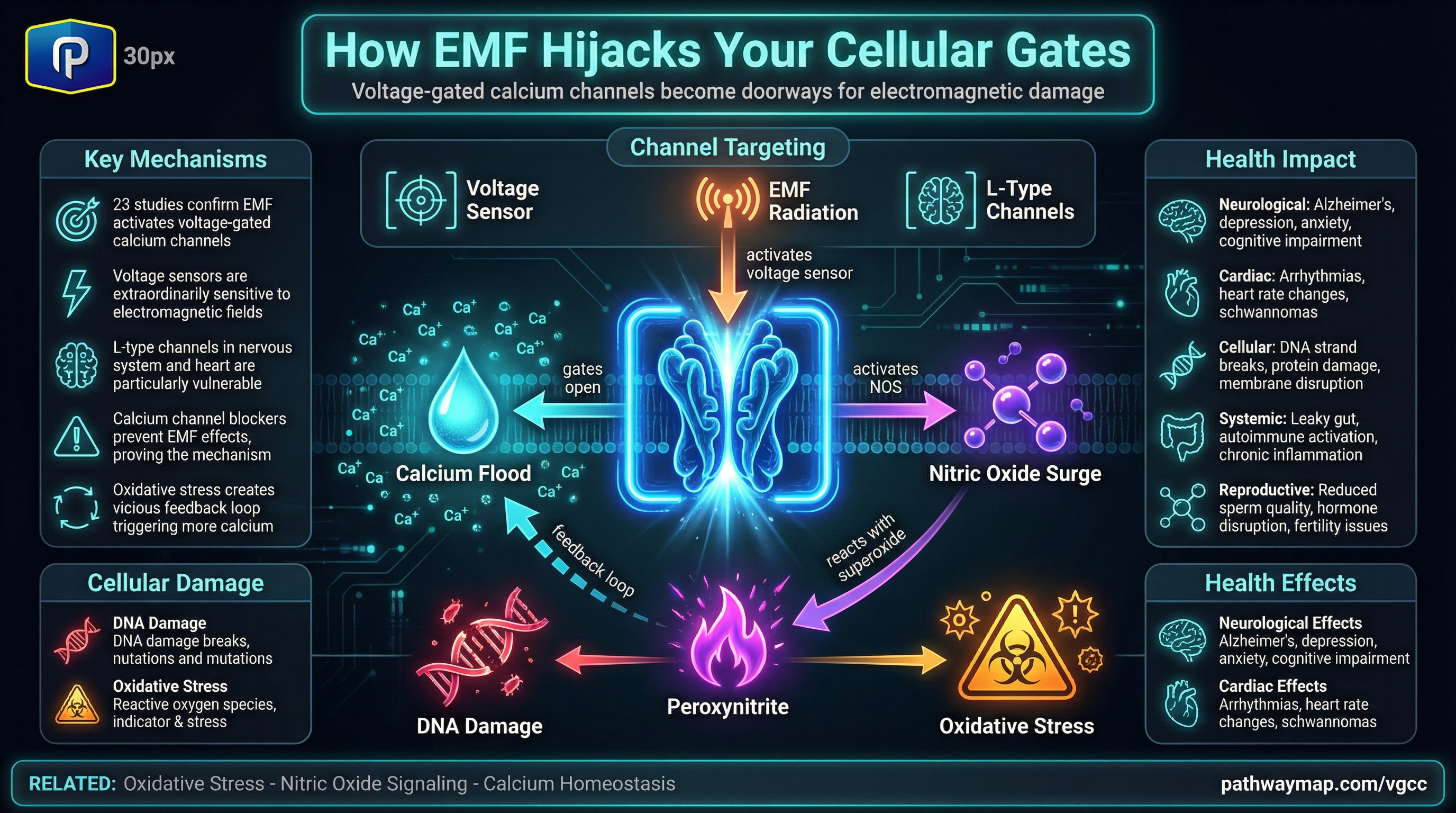
Vitamin B12 metabolism depends fundamentally on glutathione (GSH) through multiple biochemical mechanisms that extend from initial protection in circulation through final intracellular processing. This research reveals that glutathione functions not merely as an auxiliary antioxidant but as an essential cofactor determining B12 bioavailability, transport efficiency, and enzymatic function. The relationship challenges conventional approaches to B12 supplementation, particularly high-dose injections that bypass natural regulatory mechanisms.
Glutathione forms protective complexes that prevent B12 destruction
Glutathionylcobalamin (GSCbl) serves as nature’s shield for circulating B12. Research demonstrates that glutathione directly binds to cobalamin, forming a stable protective complex that resists oxidative damage far better than unprotected B12 forms. X-ray crystallographic evidence confirms this protective structure. When exposed to oxidizing agents, GSCbl maintains its integrity while other B12 forms undergo degradation.
The protective mechanism extends beyond simple shielding. Glutathione prevents B12 depletion from xenobiotic reactions by blocking the reduction of hydroxocobalamin to the highly reactive cob(I)alamin “supernucleophile.” Without glutathione’s intervention, this reactive form readily combines with environmental epoxides from industrial chemicals like chloroprene and 1,3-butadiene metabolites, permanently removing B12 from the bioactive pool. Studies show that typical physiological B12 levels may be insufficient for survival without adequate glutathione protection.
Thiolatocobalamins demonstrate superior antioxidant efficacy compared to standard B12 forms. Research reveals these compounds maintain intracellular glutathione levels and provide exceptional cell survival against oxidative stress. This dual action—protecting B12 while preserving cellular glutathione—creates a synergistic antioxidant system where each component reinforces the other’s stability.
MMACHC protein requires glutathione for B12 cellular processing
The discovery of MMACHC’s glutathione transferase activity revolutionized understanding of B12 metabolism. This protein, mutated in the most common inborn error of B12 metabolism (cblC disease), uses glutathione in a highly specific reaction that other biological thiols cannot perform. Studies reveal glutathione acts as the exclusive nucleophile for processing various B12 forms.
The molecular mechanism involves specific binding sites where glutathione interacts with the protein. Mutations at these sites disrupt GSH binding and cause severe B12 processing defects. When glutathione binds to MMACHC, it dramatically increases the protein’s affinity for B12—approximately a hundredfold improvement—explaining why glutathione depletion impairs B12 cellular uptake even when serum levels appear normal.
Glutathione enables the critical reduction of cobalt from Co(III) to Co(II), essential for generating active B12 cofactors. This process requires both reduced glutathione and FADH2, creating vulnerability under oxidative stress conditions when glutathione becomes depleted. Studies in diabetes mellitus and Alzheimer’s disease demonstrate that oxidative stress causes functional B12 deficiency through this mechanism, despite normal or elevated serum B12 levels.
Intracellular B12 enzymes depend on glutathione for function
Methionine synthase exhibits complete dependence on glutathione availability or direct provision of glutathionylcobalamin. Research demonstrates that MS utilizes GSCbl as cofactor more efficiently than other B12 forms. After oxidative inactivation, the enzyme requires either GSH-mediated cofactor regeneration or direct GSCbl provision for reactivation.
Studies in autism spectrum disorders revealed abnormal methylation capacity linked to glutathione depletion. Treatment with methylcobalamin and folinic acid significantly increased glutathione-related compounds, demonstrating the bidirectional relationship between methylation and glutathione synthesis. Neural cell studies confirm that neurotoxic metals decrease B12 enzyme activity while depleting glutathione, creating a compound effect.
Methylmalonyl-CoA mutase function indirectly depends on glutathione through oxidative stress protection. Excess methylmalonyl-CoA inhibits the methylation pathway and glutathione formation, creating a pathological cascade. Case studies in methylmalonic acidemia show severe lactic acidosis persisting until high-dose ascorbate therapy addressed underlying glutathione deficiency, highlighting the interconnected nature of these systems.
Low stomach acid creates compound deficiencies affecting both nutrients
Hypochlorhydria impairs protein-bound B12 absorption through loss of acid-pepsin digestion. Studies demonstrate that hypochlorhydric patients show markedly decreased absorption of protein-bound B12 compared to normal controls, despite maintaining normal absorption of crystalline B12. This selective malabsorption affects dietary B12 while leaving supplemental forms unaffected, explaining why serum levels may appear normal despite functional deficiency.
The gastric mucosa contains high glutathione concentrations as an antioxidative barrier, with levels particularly elevated in glandular tissue for protection against gastric acid. Research reveals that ethanol-generated free radicals affect both gastric intrinsic factor and glutathione simultaneously, with changes in B12 binding paralleling gastric GSH levels. This mechanistic link suggests oxidative stress impacts both systems concurrently, creating a self-reinforcing cycle.
Proton pump inhibitors compound the problem through multiple mechanisms. Systematic reviews confirm PPIs significantly increase B12 deficiency risk, with over half of men on long-term PPI therapy showing low B12 levels. While direct PPI-glutathione studies remain limited, research indicates these medications cause intestinal damage and adverse microbiota changes that may indirectly affect glutathione through reduced protein digestion and amino acid availability.
High-dose B12 injections can overwhelm cofactor-depleted systems
Lithium emerges as an unexpected but critical cofactor in B12 metabolism. Controlled studies found significantly lower serum B12 in lithium-treated patients compared to controls, while hair mineral analysis reveals direct associations between lithium and cobalt concentrations. Research confirms lithium involvement in B12 transport and distribution at nutritional doses, with deficiencies potentially contributing to neurological conditions.
Intensive B12 treatment creates immediate demands for multiple cofactors. Clinical guidelines document hypokalemia risk during initial therapy due to rapid erythropoiesis increasing cellular potassium uptake. The B12 Society emphasizes that therapy “puts higher demand on your body’s resources” of folate, ferritin, and vitamin B6, noting optimal levels of these nutrients as essential for therapeutic success.
Functional B12 deficiency persists despite elevated serum levels in cofactor-depleted states. Studies found no correlation between normal to elevated serum B12 and metabolic markers of deficiency. The mechanism involves vitamin B2 deficiency leading to accumulation of inactive B12 in serum, creating “Paradoxical B12 deficiency” where tissues remain deficient despite high blood levels.
B12 injections bypass critical regulatory mechanisms
Parenteral administration circumvents natural absorption controls designed to prevent vitamin imbalances. Unlike oral B12 limited by intrinsic factor capacity and minimal natural absorption rates per dose, injections deliver the full dose directly to circulation. This bypasses the intestinal barrier and the tightly chaperoned system of specific binding proteins and receptors that normally regulate cellular membrane transport.
Research demonstrates that excessive folic acid depletes serum holotranscobalamin, decreasing active B12 in circulation and limiting tissue availability. Studies of Chilean elderly receiving B12 injections found significantly lower replenishment among those with higher baseline serum folate, suggesting that folate can interfere with B12 utilization even when absorption barriers are bypassed through injection.
The glutathione requirement for B12 processing cannot be circumvented through injection. Intracellular reduction of cobalt remains essential for forming metabolically active B12, requiring both reduced glutathione and FADH2 regardless of administration route. Without adequate glutathione, B12 accumulates in inactive forms while methylation pathways remain impaired, explaining persistent symptoms despite treatment.
Conclusion
The research establishes glutathione as fundamental to B12 metabolism through mechanisms spanning protection, transport, processing, and enzymatic function. This interdependence suggests that B12 supplementation strategies must address glutathione status and other cofactors to achieve therapeutic success. High-dose injections that bypass natural regulatory mechanisms may paradoxically worsen outcomes when underlying deficiencies remain unaddressed, creating functional B12 deficiency despite elevated serum levels. The evidence supports comprehensive nutritional assessment and targeted cofactor support rather than B12 monotherapy, particularly addressing glutathione, lithium, potassium, and folate status alongside B12 replacement.