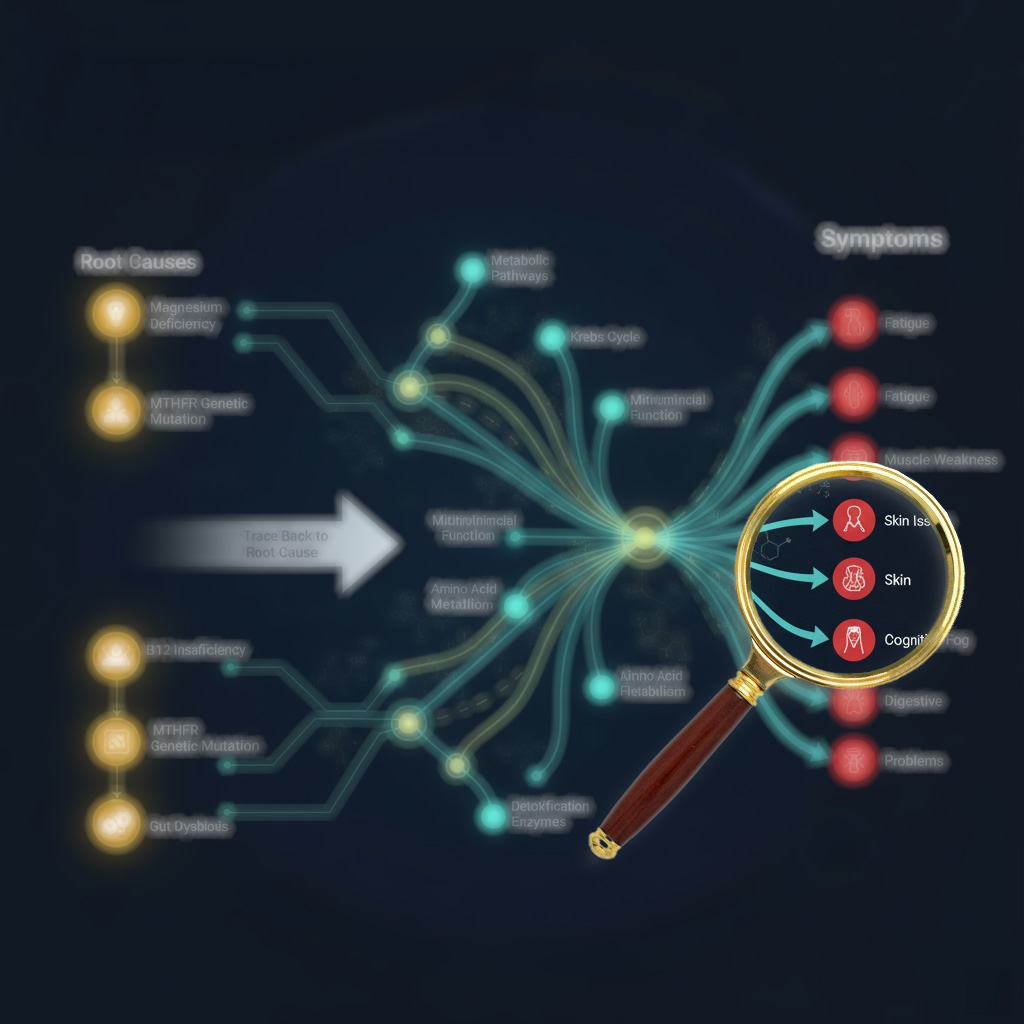
Cellular transport dysfunction unifies seemingly distinct conditions
Recent systems biology research reveals that MCAS, alpha-gal syndrome, mercury toxicity, copper imbalance, B12, glutathione disorders and so much more share fundamental mechanisms of cellular transport failure and regulatory dysfunction. Rather than treating these as separate conditions requiring different approaches, evidence suggests they represent various manifestations of the same underlying cellular crisis – where transport proteins fail, minerals become sequestered rather than deficient, and cells enter protective shutdown states that create apparent nutrient deficiencies despite adequate supply.
Cell membrane transporters fail simultaneously across multiple systems
The disruption of cellular transporters emerges as a central unifying mechanism across these conditions. Mercury directly hijacks DMT1 (divalent metal transporter 1) through molecular mimicry, simultaneously blocking iron uptake while increasing toxic metal accumulation. When DMT1 becomes upregulated due to perceived iron deficiency, it paradoxically increases uptake of mercury, lead, and cadmium – creating cascading toxicity. Anti-CD320 autoantibodies have been identified causing central B12 deficiency despite normal serum levels, demonstrating that transport failure rather than true deficiency drives symptoms. Research from 2024 shows that inflammatory conditions coordinately downregulate entire families of transporters through shared transcriptional pathways, explaining why multiple nutrient deficiencies appear simultaneously.
The copper transport system shows similar dysfunction patterns. CTR1 and CTR2 function as an interdependent system where disruption of one leads to degradation of the other through proteasomal pathways. Under inflammatory stress, CTR1 undergoes ectodomain cleavage that reduces copper transport efficiency by approximately 50%. This explains why patients can have elevated ceruloplasmin (an acute-phase reactant) while experiencing functional copper deficiency at the cellular level. Mast cell activation compounds these problems through membrane permeabilization and massive potassium efflux, disrupting the electrochemical gradients necessary for proper transporter function.
The hepcidin-ferroportin axis orchestrates mineral sequestration
Chronic inflammation from any source – whether MCAS mediators, alpha-gal immune responses, or mercury toxicity – triggers IL-6 and TNF-α release that drives hepcidin overproduction through the JAK2/STAT3 pathway. Hepcidin binds ferroportin causing its degradation within 3-6 hours, trapping iron in macrophages and enterocytes where it becomes functionally unavailable despite elevated ferritin levels. This creates the classic "anemia of chronic disease" pattern: normal to high ferritin with low serum iron and transferrin saturation.
The effects extend beyond iron. Recent research demonstrates hepcidin directly affects zinc transport by reducing ZnT1 expression via lysosomal degradation. Inflammation simultaneously increases ZIP14 expression 3.5-fold, sequestering both zinc and iron in the liver. Metallothionein expression increases up to 35-fold during inflammation, binding intracellular zinc and other metals. Studies with hepcidin antagonists show rapid mineral mobilization without supplementation, definitively proving this represents sequestration rather than deficiency. The therapeutic implications are profound – addressing inflammation rather than aggressive supplementation becomes the primary intervention.
Bile acid dysfunction creates unexpected connections
Bile acid recycling dysfunction emerges as a critical but overlooked link between these conditions. Lipophilic bile acids (chenodeoxycholic and deoxycholic acid) directly trigger mast cell degranulation at concentrations as low as 0.3 mmol/L, well within physiological ranges found in bile acid malabsorption. This creates a self-perpetuating cycle where mast cell activation disrupts bile flow, which increases colonic bile acids, triggering further mast cell degranulation.
The alpha-gal connection proves particularly revealing. Research shows that alpha-gal bound to lipids – but not proteins – crosses the intestinal epithelium and triggers systemic reactions. These glycolipids require bile acid-stabilized micelles for absorption, explaining the 3-6 hour delay characteristic of alpha-gal reactions (matching lipid digestion timing). Bile acid dysfunction simultaneously impairs fat-soluble vitamin absorption (A, D, E, K), mercury excretion through bile, and FXR/TGR5 receptor signaling that normally provides anti-inflammatory effects. Mercury compounds over 300 molecular weight are primarily excreted through bile, but disrupted enterohepatic circulation allows reabsorption, creating toxic accumulation.
Cell Danger Response explains the metabolic trap phenomenon
Dr. Robert Naviaux's Cell Danger Response (CDR) theory provides the overarching framework for understanding these interconnected conditions. When cells detect threats exceeding homeostatic capacity – whether mercury, infections, or immune challenges – they trigger CDR, shifting from normal metabolism to a defensive state maintained by extracellular ATP signaling. This creates three distinct stages: CDR1 (inflammatory containment), CDR2 (proliferation), and CDR3 (differentiation/reintegration). In chronic illness, cells become stuck in CDR1 or CDR2, unable to progress to healing.
During CDR, mitochondria fragment and adopt rounded morphology with stiffer membranes, impairing oxidative phosphorylation. Cells prioritize survival over normal function, shutting down nutrient transport and biosynthesis. This creates the "metabolic trap" – apparent B12, folate, and mineral deficiencies despite adequate serum levels. Methylation deliberately becomes impaired to prevent pathogen replication, explaining why B12/folate supplementation often fails or worsens symptoms. Suramin trials demonstrate that blocking purinergic signaling allows cells to exit CDR and restore normal metabolism, with improvements lasting 5-6 weeks after a single dose in autism patients.
Pseudodeficiency patterns reveal transport failure, not true deficiency
The distinction between absolute deficiency and functional "pseudodeficiency" fundamentally changes treatment approaches. Up to 45% of patients with functional B12 deficiency show normal serum B12 levels, with elevated methylmalonic acid revealing the cellular dysfunction. Iron pseudodeficiency presents as ferritin levels exceeding 500 ng/mL with transferrin saturation below 20% – iron is present but sequestered. Vitamin D resistance requires supraphysiologic doses because of receptor dysfunction, not deficiency.
These patterns arise from chronic stress and HPA axis dysfunction. Cortisol disrupts vitamin D activation pathways, inflammatory cytokines block nutrient utilization, and oxidative stress impairs cellular processing. Aggressive supplementation can overwhelm already dysfunctional transport systems through competitive inhibition (zinc blocking copper), transport saturation, and increased oxidative stress. Research shows that addressing root causes – reducing inflammation, supporting HPA axis function, and restoring transport protein expression – proves more effective than high-dose supplementation.
Zonulin and intestinal permeability create systemic effects
Dr. Alessio Fasano's research on zonulin reveals how gut barrier dysfunction propagates systemically. Zonulin, the only known physiological modulator of tight junctions, increases both intestinal and blood-brain barrier permeability within 60 minutes of release. Triggers include bacterial exposure, gliadin/gluten (binding CXCR3 receptors), and notably, mercury – which causes significant downregulation of claudin-1, occludin, ZO-1, and JAM1 expression.
The systemic implications extend far beyond nutrient absorption. Mast cell chymase directly increases intestinal permeability, with bacterial overgrowth triggering zonulin-mediated barrier breakdown. In alpha-gal syndrome, only lipid-bound alpha-gal crossing compromised tight junctions triggers IgE-mediated reactions. Mercury creates a positive feedback loop – damaged barriers enhance mercury absorption, which further damages tight junctions. Elevated zonulin correlates with cognitive decline in mild cognitive impairment patients who convert to dementia, demonstrating effects on the blood-brain barrier.
Therapeutic implications shift from supplementation to systems restoration
This unified understanding fundamentally changes therapeutic approaches. Rather than addressing individual deficiencies through supplementation, treatment must focus on:
(To help highlight how if we turn this off, it helps. We want to do this by fixing our environment though) Restoring cellular transport function through anti-inflammatory interventions, with IL-6 antagonists like tocilizumab successfully reducing hepcidin and improving mineral status. Hepcidin antagonists such as PRS-080#22 show promise in Phase I trials, mobilizing sequestered minerals without supplementation. Larazotide acetate, a zonulin inhibitor, has completed Phase III trials with excellent safety profiles, reducing gut permeability and systemic inflammation.
Addressing Cell Danger Response requires removing triggers (heavy metals, infections, allergens), supporting mitochondrial function with phospholipid repair and targeted antioxidants, and potentially using antipurinergic therapy to interrupt danger signaling. The focus shifts from forcing nutrients into cells to allowing cells to exit defensive states naturally.
Modulating bile acid metabolism through FXR/TGR5 agonists, bile acid sequestrants to reduce mast cell activation, and microbiome support to optimize bile acid transformation. Probiotics like Lactobacillus and Bifidobacterium not only improve bile acid metabolism but also reduce mercury toxicity through direct sequestration and barrier restoration.
Systems biology approaches reveal the interconnected nature
Recent metabolomics studies demonstrate these conditions share characteristic pathway disruptions – reduced oxidative phosphorylation, altered amino acid metabolism, impaired methylation markers, and elevated oxidative stress indicators. Systems biology modeling across 693 human gut microbial genomes now enables prediction of bile acid biotransformation potential and personalized therapeutic targets. The research shows inflammatory conditions cause coordinate downregulation of multiple transporter families through shared pathways, while toxic exposures create persistent epigenetic changes affecting transporter gene expression.
The convergence of evidence from multiple research streams – cellular transport studies, inflammatory mediator research, metabolomics, and systems biology – confirms these aren't separate conditions but different manifestations of the same underlying cellular crisis. When cells detect persistent threats, they enter protective states that create apparent deficiencies through transport failure and metabolic shutdown. Successful treatment requires addressing the root causes driving cellular dysfunction rather than attempting to override protective mechanisms through aggressive supplementation. This paradigm shift from deficiency-based to transport/utilization-based treatment offers new hope for patients with these complex, interconnected conditions.